Research
Battery Materials Discovery :
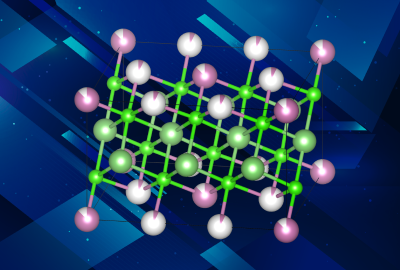
Cathode material discovery involves the employment of the Density Functional Theory (DFT) via packages such as VASP integrated with machine learning to concoct some promising candidate materials that can be used as cathodes in Na/Li-ion batteries. Our group has addressed the unique problem of predicting the properties of non-stoichimetric compounds. The work involves running DFT simulations to calculate crystal structures, stability, etc. At the same time, the ML model is trained on data acquired from sources such as Materials Project and ICSD. Using ML reduces the time and resources required.
Machine Learning & Neural Networks :

Our research group specializes in creating cutting-edge predictive models using advanced machine learning techniques and neural networks. These models enable us to predict key material properties with remarkable accuracy, which plays a crucial role in discovering novel materials for batteries. Our regression models have accurately predicted average voltage performance in various materials. In parallel, our generative neural network models are yielding promising results, showing great potential for accelerating the discovery of novel materials.
Machine Learning Force Fields :
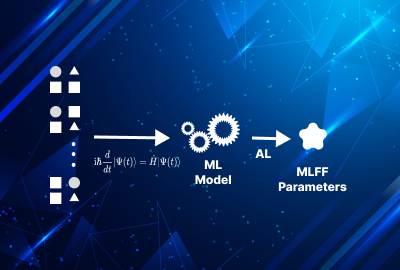
Our group utilizes machine learning force fields, with a particular emphasis on Moment Tensor Potentials (MTPs), to enhance the accuracy and efficiency of atomistic simulations. MTPs allow us to model interatomic interactions with high precision, bridging the gap between computational speed and quantum-level accuracy. These force fields are especially useful in studying complex material systems where traditional methods fall short. By integrating MTPs, we can perform large-scale molecular dynamics simulations, enabling the prediction of material behavior under various conditions, from mechanical properties to phase transitions.
Electronic Structure Studies :
The study of electronic structure, including the distribution of electronic states and spin configurations, is crucial for understanding the behavior of battery materials such as cathodes and solid electrolytes. A material's suitability for energy storage relies heavily on its electronic properties, such as the presence of a band-gap, which determines ionic conductivity and stability. Through advanced Density Functional Theory (DFT) calculations, we analyze electronic density, band structures, and charge transfer, ensuring that materials predicted by neural networks are viable candidates for battery applications with optimal performance and stability.
Reaction Dynamics Studies :

To better predict the efficiency and lifespan of an electrochemical cell, a deep understanding of reaction dynamics, including interface reactions and site-specific processes, is essential. Computationally, we achieve this by conducting extensive molecular dynamics (MD) simulations. These simulations, both classical and Ab initio, allow us to model the interactions and transformations at the atomic level. By analyzing the resulting distribution curves, we can gain insights into the reaction pathways, energetics, and progression of chemical reactions, enabling us to make accurate predictions about the performance and stability of battery materials and interfaces.
Presented Posters :



